The European Union (EU) Good Manufacturing Practice (GMP) for Medicinal Products for Human and Veterinary Use – Annex 1, commonly referred to as GMP Annex 1, was originally published as a draft in 2017. This draft left many people operating in the pharmaceutical industry wondering at some of the proposed changes to the regulations that seemed to lack details and completeness.
Now, GMP Annex 1 has been published with explanations to fill the gaps. This update is expected to not just change the EU’s guidelines, but also the US FDA’s regulations. We should expect to see the ramifications of this update throughout the world. This new update came out on the 25th of August 2022.
The major changes in Annex 1 revolve around an infrastructural approach to Quality Risk Management (QRM) through a detailed and extensive Contamination Control Strategy (CCS). We see this in heightened restrictions around movement between clean zones, personnel presence, and documentation requirements. In general, this will require extensive – and potentially expensive – adjustments for operations using older equipment that won’t be able to keep up with the new guidelines.
In this blog post, we’ll break down the first 5 parts of Annex 1’s 10 sections. We will offer a brief overview of each section to provide a basic understanding of the new regulations. In part 2, we’ll cover the remaining 5 parts.
GMP Annex 1 Part 1
Part 1 defines the scope and intent of the document. Annex 1 is designed to address the “design and control of facilities, equipment, systems and procedures used for the manufacture of all sterile products applying the principles of Quality Risk Management (QRM)”. The authors do note that, while this was written for the purpose of setting standards for sterile products, there are sections which are simply good practices for any cleanroom.
GMP Annex 1 Part 2
Part 2 is titled “Principle” and discusses the foundation of the ideology behind Annex 1. Essentially: sterile goods should be produced in an environment designed for sterility by a highly trained staff using conscious, traceable processes.
While simple in theory, this portion is where we start to see the new theme of Annex 1: automation to avoid contamination wherever possible, including removing personnel from the manufacturing line.
Humans are a major source of contamination in cleanrooms – that does not come as a surprise. So it makes sense to rely more heavily on machines and automation, but with those technological advances comes more regulations and guidances surrounding documentation, validation, safety, data integrity, and traceability.
GMP Annex 1 Part 3
Part 3 dives into the Pharmaceutical Quality System (PQS). It outlines the foundations of a good PQS:
- It should be integrated into all stages of the product life cycle.
- The manufacturer should be knowledgeable on the product in such a way that they could recognize when something went awry and put the sterility at risk.
- If equipment fails, a proper investigation should be conducted which ends with corrective and preventive actions (CAPA).
- Risk management is strictly applied and followed to avoid all types of contamination.
- Senior management should be involved in monitoring and regularly reviewing the risk management strategies.
- Finishing, storing, and transporting the product should not put it at risk.
- Those who are responsible for validating and distributing the product should have access to production and quality information. They should also have knowledge of the process in such a way as to recognize when something has gone wrong that could impact the quality of the final product.
GMP Annex 1 Part 4
Part 4 outlines the expectations for the design of cleanrooms, including the use of Restricted Access Barrier Systems (RABS), airlocks, filters, and change rooms. This section also dives into how the cleanroom should be designed, such as the materials used, ceiling shape, etc. With the introduction of additional technologies, such as RABs and isolators, to assist with the contamination control strategy (CCS), we see another one of Annex 1’s major changes.
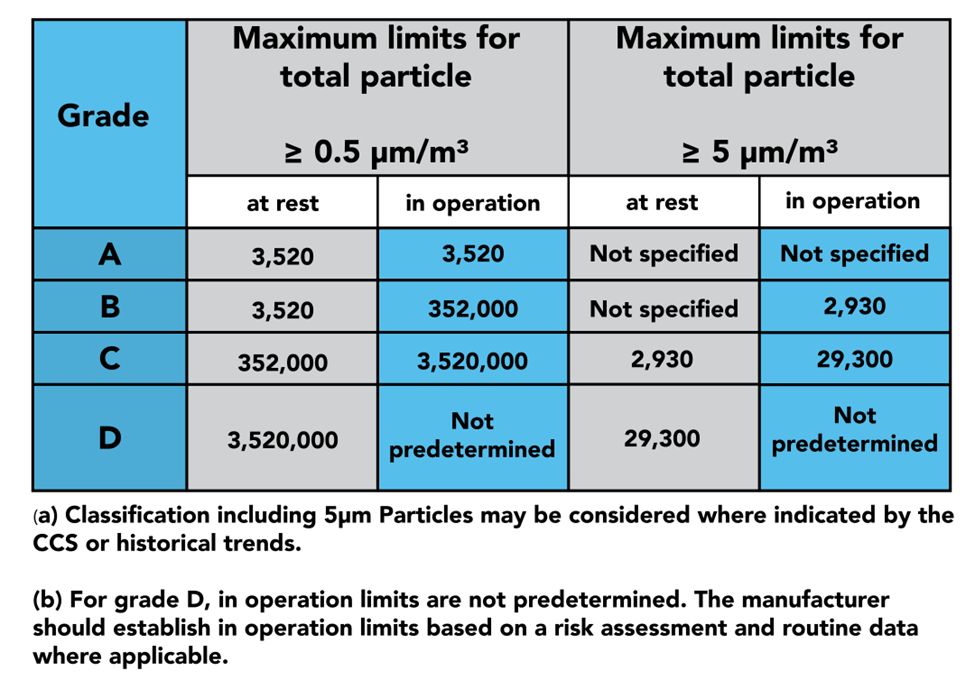
In part 4, we see the 4 grades of clean zones introduced: Grades A, B, C, and D. Grade A is considered the cleanest portion of the room, being used for highly sensitive processes, such as aseptic filling. Grade B zones are used as background rooms for Grade A zones: essentially, further separation from Grade C and D. Grade C and D zones are used for less critical stages of the manufacturing processes.
When designing your cleanroom, you should consider how objects, personnel, and air is passed from zone to zone, as this is a major source of contamination. Annex 1 Part 4 outlines how to conduct these processes in a way that minimises this risk. This is done through the use of RABS, isolators, airlocks, filters, positive and negative pressure rooms, and more.
The latter half of part 4 dives into the qualification and verification of the zones and the equipment used to monitor them. GMP dictates that qualification should be done both at rest and in operation, as demonstrated in the table above. This is where you will find instructions on sample sizes and locations to properly certify your cleanroom.
GMP Annex 1 Part 5
Part 5 lets you know how to describe and document your equipment and processes appropriately. It also addresses the need for a particle counter that is qualified.
This portion also dictates the importance of and proper cleaning of equipment used, as well as how equipment should be used between zones. For example, conveyor belts should not pass between zones, as that poses a risk of contamination.
All details available on gov official website and Annexure download bellows link
https://health.ec.europa.eu/system/files/2022-08/20220825_gmp-an1_en_0.pdf